
Use Splice aware aligner, Tophat2 to align short reads¶
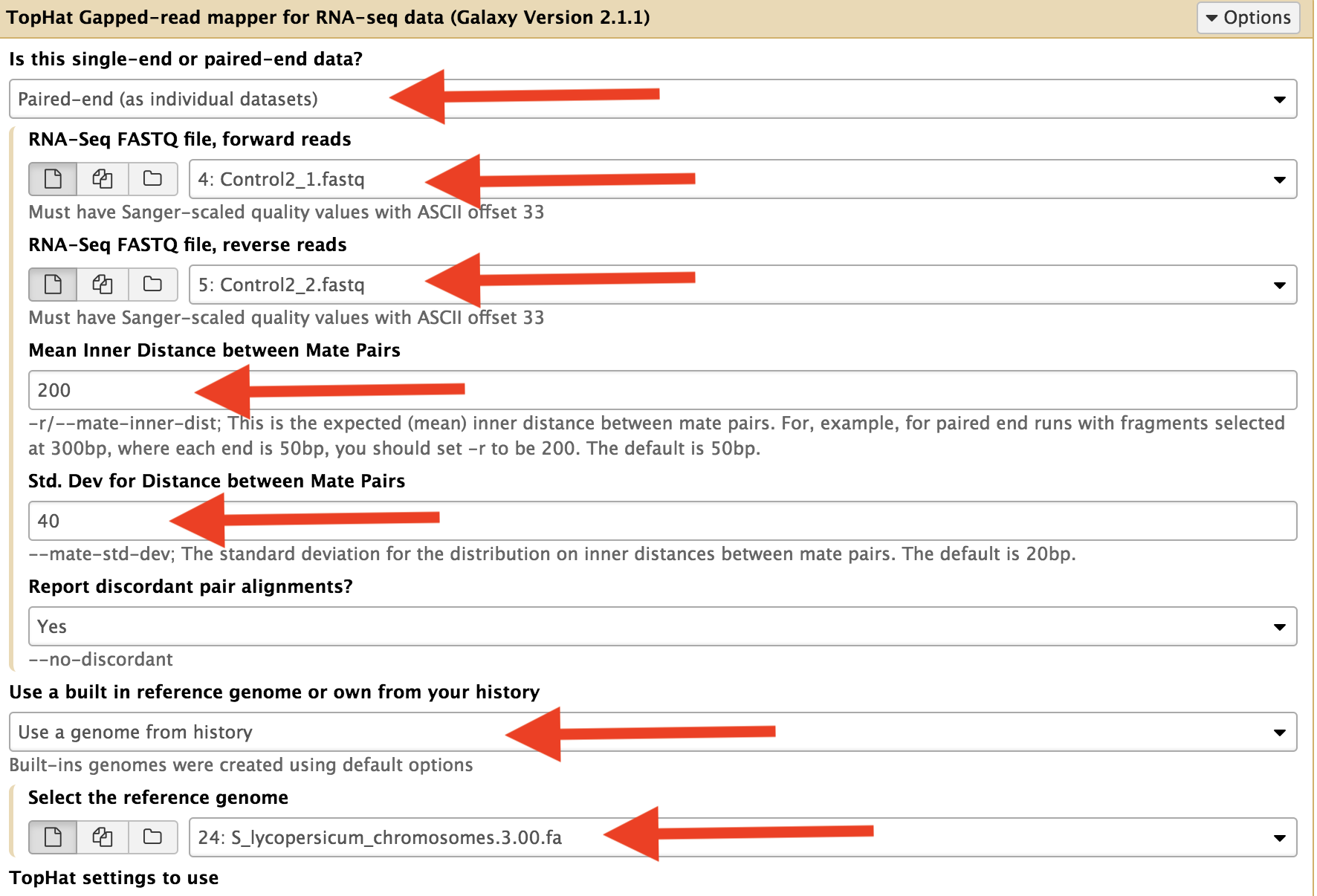
We are using Paired end reads and set the “Is this library mate-paired?” pulldown to “Paired-end”, then a second pulldown will appear to specify the 2nd FASTQ.
“Mean Inner Distance between Mate Pairs” value for this parameter should obtained from the person incharge of the sequencing.
Mean Inner Distance between Mate Pairs = length of the Fragments used for sequencing – (Length of Illumina adapters (often 120bp) + part sequenced (76+76))
Genome should be obtained from the SolGenome.net (ftp://ftp.solgenomics.net/tomato_genome/assembly/build_3.00/) and select it from the history.
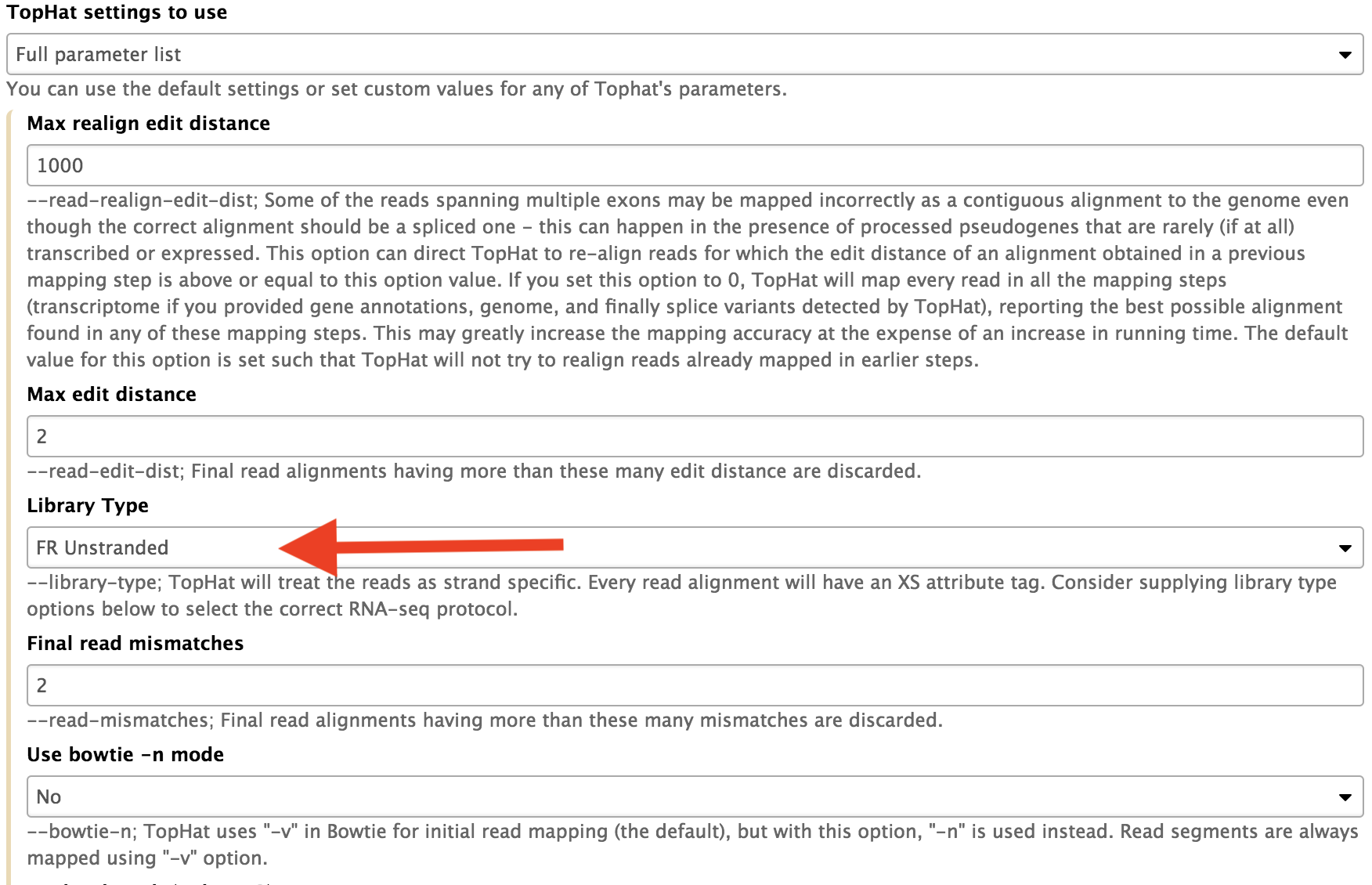
This library has been prepared to preserve the strandedness of the RNAs.
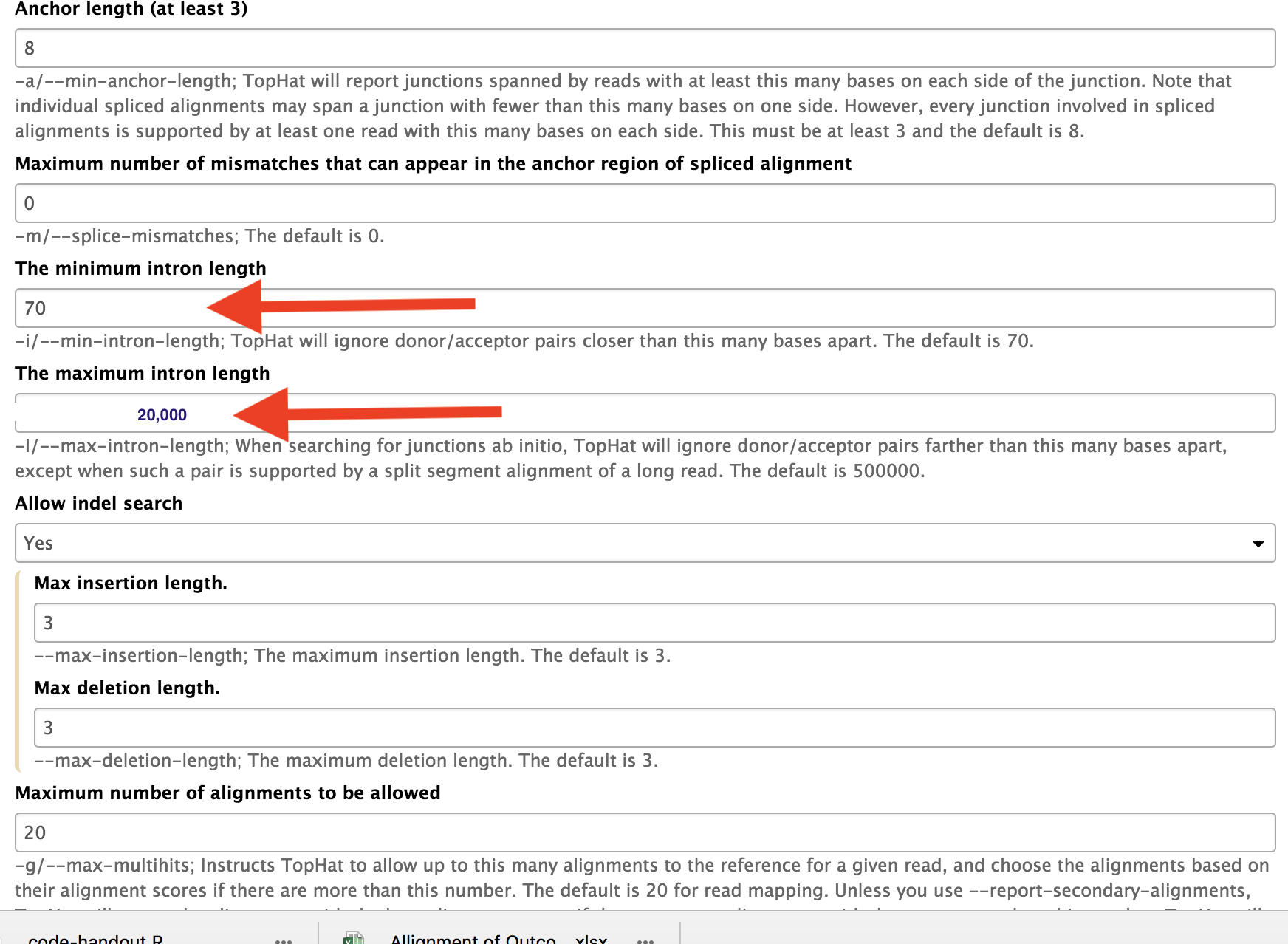
Minimum and maximum intron lengths should be changed according to genome used.
Change the intron lengths for split reads as well.
Output files:¶
- accepted_hits (BAM, BAI)
- Two binary files: .BAM (data) and .BAI (index)
3. These are the actual paired reads mapped to their position on the genome, and split across exon junctions. This can be visualized in IGV, IGB or UCSC, but you must download both .BAM and .BAI files to the same directory. splice_junctions (BED)
- BED file (list of genomic locations, no sequence) listing all the places TopHat had to split a read into two pieces to span an exon junction. This can be visualized at UCSC or in IGV, etc.
- deletions (BED) (if indel search is on)
- insertions (BED) (if indel search is on)